| Ok(n)o wiedzy genetycznej - nauka, nadzieje, kontrowersje, aktualności, ciekawostki, kontakty |
 |
 |
| Zespół Ushera |
Artykuły, publikacje | Ciekawostki | Geny z humorem | Listy do RETINY | Linki | Słowniczek
Strona główna
Spis treści Co to jest Zespół Ushera? Jaka jest szansa zachorowania na Zespół Ushera? Rozpoznanie, szanse leczenia, profilaktyka Opieka na dzieckiem chorym na Zespół Ushera Co to jest Zespół Ushera?Zespół Ushera (Usher Syndrome) jest schorzeniem dziedziczonym autosomalnie recesywnie. Charakteryzuje się głównie: uszkodzeniem słuchu typu odbiorczego oraz zwyrodnieniem barwnikowym siatkówki (retinitis pigmentosa). Oprócz tego mogą występować jeszcze inne, poważne zaburzenia.Obecnie rozróżnia się cztery typy choroby:
Należy wyraźnie zaznaczyć, że występowanie jednocześnie dwóch symptomów: schorzenia siatkówki i ubytku słuchu, nie jest jednoznaczne z wystąpieniem Zespołu Ushera. Połączenie tych dwóch schorzeń występuje dosyć często w innych rodzajach zespołów chorobowych. Aby jednoznacznie zdiagnozować Zespół Ushera, konieczne jest przeprowadzenie bardzo dokładnych badań genetycznych. Umożliwiają to nowoczesne techniki cytogenetyczne i molekularne, identyfikujące nosicieli chorób genetycznie uwarunkowanych. Dzięki nim możliwe są także: weryfikacja rozpoznania klinicznego oraz diagnostyka prenatalna. Jaka jest szansa zachorowania na Zespół Ushera?Badania molekularno-genetyczne, prowadzone wśród rodzin obciążonych Zespołem Ushera, umożliwiły dotychczas zlokalizowanie siedmiu mutacji genowych (uwaga autora - informacja ta dotyczy połowy 2002 roku), warunkujących rozwój tego schorzenia. Ocenia się, że 1 osoba na 70 jest nosicielem genu powodującego ten Zespół. Aby zachorować, konieczne jest otrzymanie zmutowanych genów zarówno od matki, jak i od ojca (szansa 1:4). W wypadku otrzymania tego genu od jednego z rodziców, nie choruje się, lecz można stać się nosicielem (szansa 2:4). Możliwe kombinacje przedstawia diagram.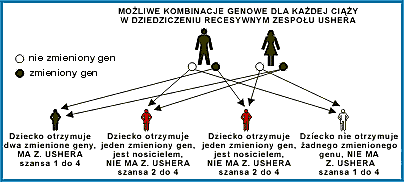 Przeważnie rodzice dowiadują się, że są nosicielami choroby dziedziczonej recesywnie przy rozpoznaniu schorzenia u pierwszego dziecka. Mimo, że prawdopodobieństwo narodzin chorego dziecka wynosi w tym wypadku 25 procent, często okazuje się, że ryzyko to jest bardzo duże. Każde kolejne dziecko może być chore, gdyż - jak mówi stare porzekadło - „los nie zna pamięci". Częstotliwość występowania zachorowań recesywnych (w tym też Zespołu Ushera), uzależniona jest ściśle od współczynnika spokrewnienia danej populacji. W hermetycznych, małych środowiskach, gdzie występują liczne małżeństwa tzw. „dalekiego spokrewnienia", szansa wystąpienia tego typu zaburzeń genetycznych jest znaczna. W miarę postępujących przemian kulturowych i spadku przeciętnego współczynnika spokrewnienia, zmniejsza się radykalnie częstość występowania chorób recesywnych. Rozpoznanie, szanse leczenia, profilaktykaNa świecie prowadzone są prace nad precyzyjnym określeniem mutacji genowych odpowiedzialnych za Zespół Ushera. Szybki rozwój genetyki molekularnej umożliwi w najbliższej przyszłości dokładne badania genotypu każdego człowieka. W ten sposób będzie można przewidzieć, czy przyszli rodzice są, czy też nie nosicielami genów, które w określonej kombinacji mogą być odpowiedzialne za, jak to ma miejsce w Z. Ushera, schorzenie recesywne swojego dziecka.Dzisiejsza medycyna nie zna skutecznych metod leczenia ludzi chorych na ten zespół. Szukanie rozwiązań dotyczących powstrzymania degeneracji siatkówki Retinitis Pigmentosa, będą miały prawdopodobnie zastosowanie także w Z Ushera (przeszczepy, leki powstrzymujące utratę wzroku itp.). Należy tutaj pilnie śledzić postęp medycyny w tym zakresie, szczególnie genetyki molekularnej. Inne schorzenia (utrata słuchu, zaburzenia neurologiczne, itd), wymagają badań naukowców z innych specjalności. W tego typu zespołach jest to największy problem - mnogość problemów, przed jakimi staje świat medycyny. Dlatego też tak ogromną wagę przykłada się obecnie na profilaktyczne zapobieganie tej chorobie. Wiedza przyszłych rodziców, co do możliwości ewentualnego wystąpienia schorzenia dziedzicznego u dziecka, stwarza szanse świadomego wyboru w miejsce "ruletki szczęścia". Być może, w niedalekiej już przyszłości, inżynieria genetyczna będzie w stanie leczyć takie, zmutowane geny już "w zarodku", przed narodzinami dziecka? Opieka na dzieckiem chorym na Zespół UsheraWczesne rozpoznanie tego schorzenia jest łatwiejsze, niż to ma miejsce w wypadku diagnozowania zwyrodnienia barwnikowego siatkówki (RP). Każde dziecko, które ma uszkodzenie słuchu typu odbiorczego, powinno być zbadane pod kątem ewentualnego wystąpienia Zespołu Ushera (w wypadku samego RP, objawy mogą być wychwycone często dopiero w drugiej lub nawet w trzeciej dekadzie życia). Tym bardziej, że obok zaburzeń słuchu i wzroku mogą występować również inne, bardzo poważne schorzenia, mające wpływ na dalszy rozwój dziecka.Dziecko z Zespołem Ushera wymaga szczególnie troskliwej opieki specjalistycznej już od samego początku rozwoju. Dotyczy to zarówno opieki medycznej, psychologicznej, jak i oświatowej oraz socjalnej. Na świecie prowadzone są ośrodki, zajmujące się tego typu dziećmi, jak choćby słynny Boys Town National Research Hospital w North w stanie Omaha w USA (adres internetowy zamieszczony został na końcu artykułu). W tym ośrodku - w roku 1998 - grupa lekarzy pod kierunkiem dr. Kimberlinga zidentyfikowała gen odpowiedzialny za Zespół Ushera Typ II - USH2A. W Europie bardzo zaawansowane badania prowadzone są przez grupę naukowców kierowaną przez prof. Petit w Instytucie Pasteura we Francji. Powoli rysują się nadzieje na pełne rozpoznanie tej choroby. Niepokojące są natomiast ostatnie doniesienia o odkryciu kolejnych mutacji genetycznych Zespołu Ushera, wynikających prawdopodobnie z ogólnoświatowej migracji ludności i tworzenia się nowych „mieszanek genetycznych". Jak widać globalizacja ma swoje, całkiem nieoczekiwane, negatywne strony. Jednak w dużych aglomeracjach, w wypadku schorzeń dziedziczonych recesywnie, szansa zachorowania jest o całe niebo mniejsza niż w małych, hermetycznie zamkniętych społecznościach, szczególnie tam, gdzie występuje wiele małżeństw spokrewnionych. Gdyby ktoś chciał podzielić się z innymi na łamach RETINY FORUM swoimi doświadczeniami, wiedzą, uwagami na temat Zespołu Ushera - to proszę napisać. Każda informacja będzie cenna, szczególnie dla osób poszukujących informacji i porady w tym zakresie. Tekst i ilustr.: PSK Źródła: 1. "What is Usher Syndrome?", Genetics Department, BTN Research Hospital, adres internetowy: http://www.boystown.org/Btnrh/genetics/usher.htm 2. "Badania molekularno-genetyczne i prognozowanie w rodzinach obciążonych zespołem Ushera", M. Krajewski, W. Skowroński, A. Salata, Klinika Okulistyczna Wojskowego Instytutu Medycyny Lotniczej w Warszawie, Kw. Medyczny "Okulistyka" Wyd. specjalne, W-wa 1999 |

Zajrzyj koniecznie do działu Nauka - aktualności.
Zamieszczane są tam najnowsze informacje dot. eksperymentów i badań naukowców z całego świata, rokowań w leczeniu chorób na tle genetycznym, profilaktyki zdrowotnej i in.

| Do Strony Głównej | Powrót | ||||
© Copyright by RETINA FORUM 2001-2016
redaktor naczelny: Piotr Stanisław Król
RETINA FORUM - Internetowe ok(n)o wiedzy genetycznej